



CO�任�����Ǻϳɰ��ȹ�ҵ����Ҫ������70����ǰ����Ӧ�õ���Fe-Crϵ���±任��������ʮ������Ȼ����ԡ������Եȵõ��˺ܴ�չ������Ŀǰ��Ȼ���ڻ����¶ȱȽϸ�(һ����300�����)���������ܲ���������к���ȱ�㣬���ֿ�������Cu-Znϵ���±任�������ô������нϺõĵ��»��ԣ����������Ż�������խ������Ͱ��ȶ���ʮ������֮ȱ�㡣Ϊ�˽����Щ���⣬��������Co-Moϵ��������任����������Щ�������������������о����շ��Ʊ�����������Ľṹ���������������ִ�������������������ԣ����Ҿ��кܿ��Ļ������������ȫ�������Fe-Crϵ��Cu-Znϵ�任�����IJ��㡣��
�������ͨ���Ԧã�Al2O3�����壬�����Գɷ�Co��Mo��Ni��W�����ò�ͬ���Ʊ��������ص������ϡ�Ϊ�˸������弰������ijЩ���ܣ����������������Al2O3+MgO��Al2O3+Re2O3��Re����ϡ��Ԫ�أ����Լ����Ӽ������������Ҳ�б�������
�������͡����ͻ�úΪԭ������ϳɰ�ԭ����ʱ��ʹ��Co-Moϵ���±任�������Խ���������ֱ�ӽ��б任���پ��������̼�������̣��������ؽ������ܺġ���˽���ʮ������Co-Moϵ���±任�������о����ر��ǹ�ҵӦ�ü�ʹ�ü������о��������ϵõ���Ѹ�ٵķ�չ����
1 Co-Moϵ�����Ľṹģ����
1.1 ����ã�Al2O3�Ľṹ��
�ڹ�ҵ����������������;�㷺�����壬����1797��������Ծ��ѱ��˷��֡��ã�Al2O3�����ķ���ϵ���侧�������ڼ⾧ʯMgAl2O4�Ľṹ���ڦã�Al2O3�У���21.5����ԭ�ӷֲ���24�������Ӳ�λ���ڰ�����λ������2 (2/3)����λ����8�������ӷֲ����������϶�ڣ��൱��A (1/8)��A113 (1/2)��2 (2/3)��O32�Σ����пڱ�ʾ��λ���ã�Al2O3���нϴ�ıȱ��漰������ġ�Rosynek����Ϊ���ã�Al2O3�������ֲ�ͬ�Ŀ�Ѩ����Щ��Ѩ�����OH������һЩ��Ҫ�Ĺ�ҵ�����������һ�ֻ��Խ��棬��ͬ�����������Ų�ͬ�����á���
1.2 Mo���ã�Al2O3�Ľṹ��
Payen�������������о������չ���Mo���ã�Al2O3��������Ϊ���ص�����Ҫ��Mo7O246-��ʽ���ڣ�������������ṹ�Ǻ��ȶ��ģ������γ���Mo-O-Al��������������Ŀ�����Hall�����ú�������о�����ļ���Ԧã�Al2O3�������-OH��Ӱ�죬���Ƿ���Mo���ã�Al2O3����-OH�״����⸺���������Ӷ�����ʧ���������������ã�Al2O3����������á���ͼ1��ʾ��
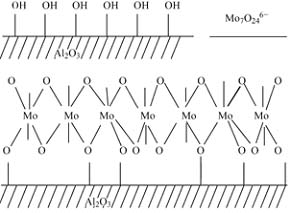
ͼ1 �����-Al2O3�����ʾ��
1.3 ����MoS2/�ã�Al2O3�Ľṹ��������ģ��
Massothָ��������������̬�����̬�Ժ�������ṹ���ֲ��䣬ֻ����ԭ�Ӳ��ֵ��������ԭ�ӵ�λ�ã�����һС������ԭ�ӱ�H2��ԭ�������˿�λ����Щ�ֲ��䡣��ͼ2��
ͼ2 ����ģ��ʾ��ͼ
���ģ����Ҫ����Mo/�ã�Al2O3�ṹ��չ�����ģ�ȱ��Co2+��K+���о����ݡ���
1.4 ����ģ�ͪ�
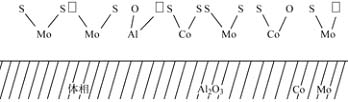
����Խ��Խ�������Ϊ���ԸߵĴ�����Co�����Զ�����������ڣ�Co���Զ�λ�ڲ�״����IJ�ͬ����λ���ϣ���ͼ3��ʾ��

ͼ3 MoS2������Co����λ�ṹʾ��ͼ
Frrangher����Ϊ��λ��MoS2�ı�Եλ��b����λ�ڲ��aλ���Ӷ�ʹ�������λ��������������¶����������˻��ԡ�Co���ı�Mo���Ե�Ļ��ԣ�ֻ�����˻���λ��Ŀ�����˽ṹ���������á�����ģ����任��Ӧ����֮��Ĺ�����δ����������
1.5 �Ӵ�Эͬ����ģ�ͪ�
Delmon��Ϊ��������������Ŷ�����Co9S8��MoS2�࣬�����ĽӴ�ʹCo9S8������MoS2�ṩ����������������á�Ȼ����һģ���ڽ���HDS���������Co/Mo��֮��Ĺ�ϵʱ�������鷳����
1.6 Co-Mo-Sģ�ͪ�
�Դ�Tops��e���������Co-Mo-S��ģ�ͺõ���Խ��Խ��ʵ������֧�֡������ֽṹ��Co��λ��������MoS2�����б�Moԭ�ӵ�λ�ã���ͼ3�е�dλ��ʵ��֤��Co-Mo-S����Coԭ�ӵĶ�������HDS���Եĸߵ��кܺõ����Թ�ϵ��Eltznel����ʵ��Ļ����ϣ�����Co-Mo-Sģ�ͣ�ϵͳ�ؼ����˵�һ���ڸ����ɽ���Ԫ����MoS2�γɵ�Co-Mo-S���Ӵص��ܼ�ͼ������ʵ������������ӵ���ת�ƽǶȸ���̵ز��������������á���
�����о���Co-Mo-S��ṹ��任����֮��Ĺ�ϵ��ȡ����һЩ�����������Ϊ��Co-Mo-S��ṹģ����ΪCo-Moϵ����任�����Ļ������ģ���ʵ�����Ľ����DZȽϺ����ġ���
2 Co-Mo����任������������
2.1 �������Ʊ���
Co-Moϵ������±任�������Ʊ����кܶ�ר�����ױ�����һ��ɷ�Ϊ��Ϸ�����շ����֡���Ϸ��ǽ������ܣ�����泥�����������ˮ��Һ���������ۼ�ճ����Ȱ��涨����Ȼ����ѹ���پ����������ѹƬ���ɡ����շ��ǽ����͵�����ֽ��������ܡ�����李������������Һ������ɱ��ն���(�������ĩ�Ƚ��ջ��������Һ�������Σ��ٺ�ɱ��ն���)�����ַ����Ƚ϶��ԣ����շ����ŵ�϶ࣺ��Ҫ�ǻ��Գɷֲַ��������ⲿ������������ʣ���������Ļ��ԣ�ǿ�ȸߣ���ˮ���ۻ������ܶ�С���Ƴ̼���Ʒ���ʸߣ�����Ⱦ�ȡ���
2.2 Co-Mo����任��������Ҫ�ص㪥
���ִ�����Fe-Crϵ��Cu-Znϵ�任�����Ƚ϶��ԣ����������ص㡣��
(1)���нϿ��Ļ������������������һ��Ϊ160��500�棬�������Ƶ�B303Q����������ҵӦ��֤������120������㹻�ı任���ԣ���˸ô�������Ϊ���±任����������Ϊ���±任����ʹ�����������ģ���Ҫ����������һ������H2S�������¶����ߣ�����������Ҫ����͵�H2S����Ҳ�����ߡ�
(2)�����õ������뿹�����ܡ���Co-Moϵ�����Ļ��Գɷ���������뽫����������������ԣ����ʿ���100g��m3���µĸ�����������ϵ��ͭпϵ�任������������ġ��������ж������ʵĿ�����Ҳ�ȽϺã�������NH3��HCN��C6H6�ȶԴ����Ļ��Ծ���Ӱ�졣��
(3)������ˮ����ѹ�����ܡ��Լ�Ϊ������Co-Mo������һ���ܽϲ����þΪ������Co-Mo������һ���ܷdz�ͻ�������ִ����Ļ�������һ����280��500�棬�ڵ�ѹ�͵��������»��Բ������룬Ȼ���ڸ�ѹ��(��3.0��8.0MPa)ʹ��ʱ�������������ã���һ���������������¿�ʹ�任¯����CO����1.0�����ҡ���
(4)ǿ�ȸߡ��������������ǿ�����0.5��1������ͨ��Fe-Crϵ������ԭ̬��ǿ�ȱ�����̬��Ҫ��Щ����
(5)��������һ���ڹ�ҵװ�������ִ�����ʹ��5�����ң�Ҳ��Ӧ��10�����ϵ����ӡ�
Ϊ��ʹ��������������ڹ�ҵװ���еõ�����Ч�������Ƕ�����������������������������о�����������Ӧ��֤���������Ĵ����������Ը���CS2���ڵ�һϵ�����⣬�����Ĵ����任����Ҫ����CS2�ã��ر��ǵ��»��ԡ���
�����ĵ��»����Ƿdz���Ҫ�ļ���ָ�ꡣ���任����CO����Ϊ3��ʱ���¶������������Ĺ�ϵ���1��ʾ��
��1 �任����COΪ3��ʱ��ͬ�¶�����������
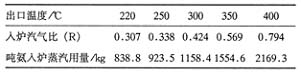 ��
�� ��Ȼ�任��Ӧ�¶����ͣ���ƽ������������������Ҳ���͡���
3 ʧ���о�
���ִ���ʧ����о������������ࡣBAFC��˾��ʹ�ù��Ĵ������й���������ΪӰ����������Ҫ�����������л����ʡ������������2��
��2 ʧ��K8-11�����ķ������
 ��
�� �ӱ�2���Կ�������Ȼ������Ӧ���еĴ���ʹ���������1�����ϣ��������������࣬��������Ļ�ѧ������Ҫ��2����ߵöࡣ�����е�̼����Ϊ�ҷ�ϴ����δ��������������������¡�����������ԭ�����еĻҷ��Ը��Ե��ʻ���������ʽNi(CO)4��Fe(CO)5�����ģ��ڻ��������ڴ������ʻ���������ֽ⣬���������ʽ������������
�����ϵȶ�������������о���ָ������Ӧ�����з���֮���������ʧ��ת������Ҫ���ܷ�Ӧ�¶ȡ������Ⱥ�ԭ��������Ũ�����߹�ϵ��Ӱ�죬����ʧ���������ʧ�����ͨ������������������Ӧ�¶�ƫ�ߺ�ˮ�������������������������������������ã����»�����IJ�����ʧ����ת�����γɷǻ�������������Σ����Dz�����ʧ�����Ҫԭ��MgAl2O4��CoS2��CoSO4���γɴ������������þ���ܵ����á���һ��������ǵ��о����һ�¡���
���Ƕ�Co-Mo��A12O3-MgO����任������ʧ�������ϵͳ�о�����X ��������������֣��ý��շ��Ʊ��Ĵ���û�й۲쵽���Գɷֵ�����壬���ù��������������Ʊ��Ĵ������Դ���MgO�Ľᾧ���࣬���ǽ��շ��Ʊ��Ĵ������»��ԽϺõ�ԭ��֮һ����
Ϊ�˽�һ���˽����ʧ���ԭ�������ֱַ��ʹ����һ�ꡢ���ꡢ���ꡢ�����Լ��ս��˵���Ʒ������XRD����(��ͼ4)����
�Ƚ�2��3��4��5�����Ľ�����Կ���������ʹ��ʱ����ӳ����������������ӣ��ã�Al2O3 ����岻����ͽǶ�λ�ƣ�������⡣��������γɵģ�gAl2O4�ᾧ���������࣬��������ڹ�ҵװ�������������ʹ��ʱ����������½���һ���Ĺ�ϵ��������λ�ƿ�����CoAl2��4��A12(MoO4)3��������йء�
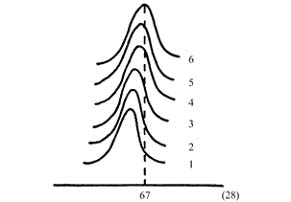
ͼ4 ��ͬʹ��ʱ��Ĵ�����Ʒ�Цã�Al2O3��XRDͼ
1��K8-11(������ṹ)��2��K8-11(ʹ��4��ṹ)��3��K8-11(ʹ��3��ṹ)��4��K8-11(ʹ��2��ṹ)��5��K8-11(ʹ��1��ṹ)��6���ã�Al2O3
�任����ѹ���Դ�������ṹ��һ����Ӱ�졣�����о��˲�ͬ���������´�������Ʒ����ͼ5��ʾ��

ͼ5 ����ͬ�������������Ĵ���XRDͼ��
1����8��Pa�²���240�죻2������1044���ս�2.5Сʱ��2��Pa�²���210�죻3����Ϊ�ã�Al2O3��4����Ϊ����Al2O3����
��ͼ5��l�����ķ���������������շ��Ʊ��Ĵ����ڸ�ѹװ��������240��ã�Al2O3�н�40����ɦ���Al2O3�����ڵ�ѹ����������210�죬���ܾ���һ�θ��´�������������û�Ц���Al2O3�γɡ���һ���˵����ѹ���Դ�������Ľṹ�����Ե�Ӱ�졣
�о����֣�Mo��ͨ��Mo-O-Mo�����������ģ������������ս��Ժ�������λ��(MoO42-)�ĺ���������������ʾ������Al2(MoO4)3����һ���֤����Co-Mo����任��������700�����ϸ����ս��Ժ�����ͻ��Գɷַ����˻�ѧ���ã������ɵĻ������Ƿdz��ȶ��ģ�����Ҳ�����ܽ������л��Ե�������Ǵ���ʧ���ԭ��֮һ����
XPS�о����ָ����Co��ʹ��ǰ��Ҫ������������ʽ���ڣ����������ս����Ʒ���������ķǻ�����CoAl2O4���ڣ������Ժ�CoO���CoS��CoAl2O4����䡣�������������Ժ����CoMoO4�������ɣ��ٴ����Ժ�CoMoO4��ɻ��Խϵ͵�CoMoS4���Ӷ���ɴ�����ʧ���
��������ICP��ʧ��ǰ��ļ���Co/Mo/MgO-A12O3������Co��Mo��Mg����Ч�ɷֽ����˷��������������ͼ6��
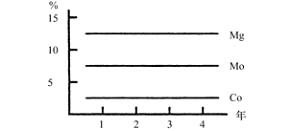
ͼ6 ʹ�ò�ͬʱ��Ĵ����л��Գɷֵ�ICP�������
��ͼ6�ɼ����������Co��Mo��Mg����Ч�ɷ��ڹ�ҵʹ�ù�����ʼ�ձ��ֲ��䡣��һ���֤�������������ʧ������һ�����ء���ҵʹ�ú�����е����ʺ�������3��
��3 �����е����ʷ������ 10-6��

�ɱ�3�ɼ�����������Ҫ��Fe��Cu�������������ߡ���Ϲ�ҵװ��֮ʵ�ʽ�������ʶԴ������Ե�Ӱ����Ҫ�ǡ����С��˴�������������Ӧ���Ĵ���������ͨ�������Ժ��������ֿɻָ���֤������һ�㡣��
4 ��������
������������Կ���������Ϊ���ã����ǶԴ˽����˴������о����������Ĺ�ҵӦ��֤������һ�����ϴ�ͳ����������ܶࡣ��ɽ�����ʳ�������Ϊ������һ���ܡ���
��ǰ�Ĵ����¶ȼ���4����
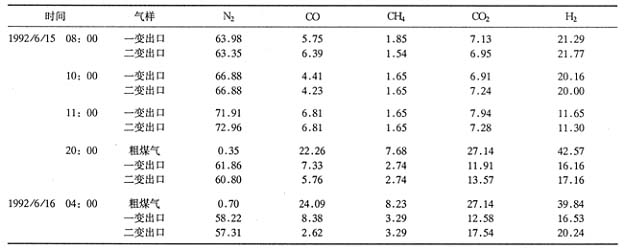
�����У�ϵͳѹ��������0.35MPa����N2����ά����6000��7800m3/h����5h��ú������������6000m3/h����6h��CS2��������Ϊ50L/h��150L/h��200L/h�������¶���26h֮��һ��¯��200������370�棻����¯��200������330�档�ô���ʹ��H2S���������ߣ���һ��¯��0.0061����ߵ�0.21��������3.5��;�ڶ���¯��0.0036��������0.018�������5�����������ݼ���5����6����7�ͱ�8��ʾ��
��4 ��ǰ�����¶� ��
 ��
����5 ������H2S������� ��

��6 ������������ʱ��Ա�
 ��
����7 ������ȫ������� ��
 ��
����8 ����ԱȪ�
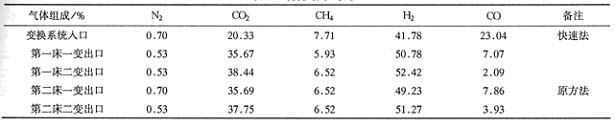
�ɱ�6��7�ɼ����·��������Ⱦɷ�����ʡ5��7��ʱ�䣬���Ҵ������Ա��Ϸ����á�
5 ����
���Ķ�Co-Moϵ����任�������Ʊ����ṹ�������ﻯ���ܡ�������ʧ���о��Ƚ����˽�Ϊ��ϸ�Ľ��ܡ���һ������ʮ���������ش�ĸ��ƣ��Ժϳɰ��Ľ��ܼ��Ƴ̵Ľ��������˻��������á�����Co��Mo�ļ۸�ϸߣ���˳��Բ��ü۸�ϵ͵�Ԫ����ȡ��Co��Mo��ʮ��������Ĺ������������Ų��õĽ������ⷽ��Ĺ�������ȡ���µ�ͻ�ơ�

Ԫ����Ѷ��
������״�������
������
������
��������
���������� (Ӣ�İ�)
��Ʒ������
VIP������ֲ�
������̳
QQȺ������
��ַ���ɶ����������������18��8�� �绰��028-83667786��83667576��89309098 ���棺028-83667578